KinoML object model¶
The KinoML object model provides access to binding affinity data in the context of machine learning for small molecule drug discovery (Fig. 1). The DatasetProvider is the central object for storing all relevant information of a dataset. It is essentially a list of Measurement objects, which contain the measured values (singlicate or replicates), associated to a System plus experimental AssayConditions. A System is a list of MolecularComponent objects; usually a
Protein and a Ligand. Featurizers will use the input of the MolecularComponents to represent the System in different formats for machine learning tasks, e.g. a Ligand as molecular fingerprint.
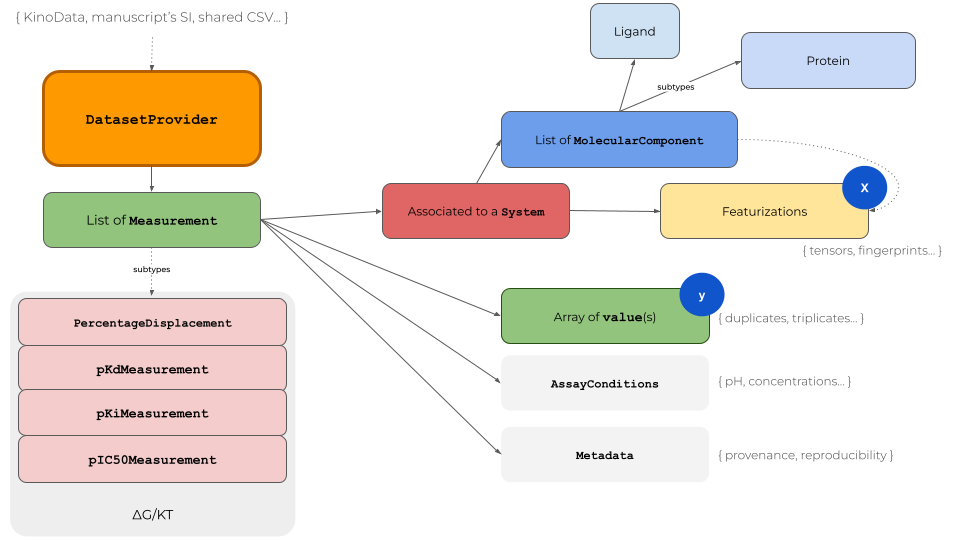
KinoML has a focus on protein kinases but the architecture is applicable to protein targets in general. When writing your own KinoML objects it is recommended to move computational expensive tasks to the Featurizer level, which is capable of multi-processing. For example Protein objects can be initialized with nothing else but a UniProt ID. The amino acid sequence will be fetched when the Protein’s sequence attribute is called for the first time. Thus, one can quickly generate
many Protein objects and the more time-consuming sequence fetching is done with multi-processing during featurization.
In the following section, different KinoML objects will be introduced including code examples.
Molecular components¶
Molecular components like ligands and proteins store molecular representations, a name and additional metadata, that may be important for working with the data and provenance.
Ligands¶
Ligand objects store information about the molecular structure of a ligand, usually a small molecule with certain activity for a target.
The Ligand object is based on the OpenFF-Toolkit Molecule object, which can be accessed via the molecule attribute. This also allows usage of methods of the OpenFF-Toolkit Molecule including conversion to other toolkits, e.g. RDKit and OpenEye. The Ligand object can be directly
initialized via SMILES or file including interpretation of the given input, or lazely initialized via SMILES without any interpretation of the given input.
[1]:
from openff.toolkit.utils.exceptions import SMILESParseError
from kinoml.core.ligands import Ligand
[2]:
# initialize a Ligand from SMILES, the molecule will be directly interpreted
ligand = Ligand.from_smiles("CCC", name="propane")
print(type(ligand))
print(type(ligand.molecule))
print(type(ligand.molecule.to_rdkit()))
print(ligand.molecule.to_smiles(explicit_hydrogens=False))
<class 'kinoml.core.ligands.Ligand'>
<class 'openff.toolkit.topology.molecule.Molecule'>
<class 'rdkit.Chem.rdchem.Mol'>
CCC
[3]:
# erroneous input will raise errors during initialization
try:
ligand = Ligand.from_smiles("XXX", name="wrong_smiles")
print("Success!")
except SMILESParseError:
print("Failed!")
Failed!
Warning: Problem parsing SMILES:
Warning: XXX
Warning: ^
[4]:
# Ligands can also be lazely initialized via SMILES
# here the interpretation is done when calling the molecule attribute for the first time
ligand = Ligand(smiles="CCC", name="propane")
print(type(ligand.molecule))
<class 'openff.toolkit.topology.molecule.Molecule'>
[5]:
# this makes the object generation faster
# but will result in interpretation errors later, e.g. during a featurization step
# hence featurizers need to detect and remove those systems
ligand = Ligand(smiles="XXX", name="wrong_smiles")
print("Ligand lazely initialized!")
try:
print(type(ligand.molecule))
print("Success!")
except SMILESParseError:
print("Failed!")
Ligand lazely initialized!
Failed!
Warning: Problem parsing SMILES:
Warning: XXX
Warning: ^
Proteins¶
Protein objects store information about the molecular structure of a protein, e.g. the target of a small molecule inhibitor.
KinoML provides two different Protein objects, i.e. Protein (applicable to all proteins) and KLIFSKinase (allows access to information from the protein kinase-specific KLIFS database). Similar to Ligand, protein objects can be directly or lazily initialized.
Again, the molecular structure is accessable via the molecule attribute. However, both protein objects support two toolkits, i.e. MDAnalysis and OpenEye, which can be specified via the toolkit argument. A conversion from one toolkit to the other after initialization is currently not possible, but likely not needed anyway.
Another important attribute of proteins is their sequence. Depending on the used featurizer, a molecular structure may actually not be required, for example in case of OneHotEncoding of the sequence. Hence, you can also initialize Protein and KLIFSKinase using sequence identifiers only, e.g. UniProt ID or NCBI ID. This is always done lazily, so the sequences will be fetched from the respective resource on the first call of the sequence attribute. Protein and KLIFSKinase
inherit their sequence-related functionality from the AminoAcidSequence object in kinoml.core.sequences, which allows for further a customization of sequences, e.g. mutations. For more details have a look at the AminoAcidSequence class in the respective section of the KinoML API documentation.
[6]:
from kinoml.core.proteins import Protein, KLIFSKinase
/home/david/miniconda3/envs/kinoml/lib/python3.9/site-packages/numpy/core/getlimits.py:499: UserWarning: The value of the smallest subnormal for <class 'numpy.float64'> type is zero.
setattr(self, word, getattr(machar, word).flat[0])
/home/david/miniconda3/envs/kinoml/lib/python3.9/site-packages/numpy/core/getlimits.py:89: UserWarning: The value of the smallest subnormal for <class 'numpy.float64'> type is zero.
return self._float_to_str(self.smallest_subnormal)
/home/david/miniconda3/envs/kinoml/lib/python3.9/site-packages/numpy/core/getlimits.py:499: UserWarning: The value of the smallest subnormal for <class 'numpy.float32'> type is zero.
setattr(self, word, getattr(machar, word).flat[0])
/home/david/miniconda3/envs/kinoml/lib/python3.9/site-packages/numpy/core/getlimits.py:89: UserWarning: The value of the smallest subnormal for <class 'numpy.float32'> type is zero.
return self._float_to_str(self.smallest_subnormal)
[7]:
# initialize from PDB ID with different toolkits
protein = Protein.from_pdb("4yne", name="NTRK1")
protein2 = Protein.from_pdb("4yne", name="NTRK1", toolkit="MDAnalysis")
print(type(protein.molecule))
print(type(protein2.molecule))
protein2
Warning: importing 'simtk.openmm' is deprecated. Import 'openmm' instead.
<class 'openeye.oechem.OEGraphMol'>
<class 'MDAnalysis.core.universe.Universe'>
[7]:
<Protein name=NTRK1>
[8]:
# initialize lazily via PDB ID
protein = Protein(pdb_id="4nye", name="NTRK1")
print(type(protein.molecule))
<class 'openeye.oechem.OEGraphMol'>
[9]:
# note there is no sequence yet, since no UniProt ID was given
print(len(protein.sequence))
# but one could get it from the protein structure if needed
0
[10]:
# initialize with sequence from UniProt
protein = Protein(uniprot_id="P04629", name="NTRK1")
print(protein.sequence[:10])
# initialize with sequence from UniProt and custom mutations
protein = Protein(uniprot_id="P04629", name="NTRK1", metadata={"mutations": "R3A"})
print(protein.sequence[:10])
print(type(protein.molecule)) # a molecule is not available
MLRGGRRGQL
MLAGGRRGQL
<class 'NoneType'>
[11]:
# get the kinase KLIFS pocket sequence via different identifiers (lazy)
kinase = KLIFSKinase(uniprot_id="P04629", name="NTRK1")
print(kinase.kinase_klifs_sequence)
kinase = KLIFSKinase(ncbi_id="NP_001007793", name="NTRK1")
print(kinase.kinase_klifs_sequence)
kinase = KLIFSKinase(kinase_klifs_id=480, name="NTRK1")
print(kinase.kinase_klifs_sequence)
WELGEGAFGKVFLVAVKALDFQREAELLTMLQQHIVRFFGVLMVFEYMRHGDLNRFLRSYLAGLHFVHRDLATRNCLVIGDFGMS
WELGEGAFGKVFLVAVKALDFQREAELLTMLQQHIVRFFGVLMVFEYMRHGDLNRFLRSYLAGLHFVHRDLATRNCLVIGDFGMS
WELGEGAFGKVFLVAVKALDFQREAELLTMLQQHIVRFFGVLMVFEYMRHGDLNRFLRSYLAGLHFVHRDLATRNCLVIGDFGMS
Systems¶
Systems store all molecular components for a given activity data point. They may only contain a Ligand in case of purely ligand-based featurization but can also contain a Protein, i.e. LigandSystem, ProteinSystem, ProteinLigandComplex.
[12]:
from kinoml.core.systems import LigandSystem, ProteinSystem, ProteinLigandComplex
[13]:
ligand = Ligand(smiles="CCC", name="propane")
protein = Protein(uniprot_id="P04629", name="NTRK1")
[14]:
system = LigandSystem(components=[ligand])
system
[14]:
<LigandSystem with 1 components (<Ligand name=propane>)>
[15]:
system = ProteinSystem(components=[protein])
system
[15]:
<ProteinSystem with 1 components (<Protein name=NTRK1>)>
[16]:
system = ProteinLigandComplex(components=[ligand, protein])
system
[16]:
<ProteinLigandComplex with 2 components (<Ligand name=propane>, <Protein name=NTRK1>)>
Featurizers¶
Featurizers ingest Systems to compute features for e.g. machine learning tasks. Systems failing during featurization will be removed, e.g. erroneous SMILES. Featurizations are stored in each system for later usage.
[17]:
from kinoml.features.ligand import MorganFingerprintFeaturizer
[18]:
# generate systems with lazily initialized ligands
systems = [
LigandSystem(components=[Ligand(smiles=smiles, name=str(i))])
for i, smiles in enumerate(["C", "?", "CC", "CCC"])
]
systems
[18]:
[<LigandSystem with 1 components (<Ligand name=0>)>,
<LigandSystem with 1 components (<Ligand name=1>)>,
<LigandSystem with 1 components (<Ligand name=2>)>,
<LigandSystem with 1 components (<Ligand name=3>)>]
[19]:
# the featurization will lead to interpretation of the given SMILES for the first time
# failing systems will not be returned
featurizer = MorganFingerprintFeaturizer()
systems = featurizer.featurize(systems)
systems
Warning: Problem parsing SMILES:
Warning: ?
Warning: ^
[19]:
[<LigandSystem with 1 components (<Ligand name=0>)>,
<LigandSystem with 1 components (<Ligand name=2>)>,
<LigandSystem with 1 components (<Ligand name=3>)>]
[20]:
# featurizations are stored in each system as a dict
# the lastly performed featurization is additionally stored with the "last" key
systems[0].featurizations
[20]:
{'last': array([0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0]),
'MorganFingerprintFeaturizer': array([0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0])}
Measurements¶
Measurements combine information for a given activity data point, i.e. System, AssayCondition and activity values. Currently available Measurement objects are PercentageDisplacementMeasurement, pIC50Measurement, pKiMeasurement, pKdMeasurement.
[21]:
from kinoml.core.conditions import AssayConditions
from kinoml.core.measurements import PercentageDisplacementMeasurement
[22]:
ligand = Ligand(smiles="CCC", name="propane")
protein = Protein(uniprot_id="P04629", name="NTRK1")
measurement = PercentageDisplacementMeasurement(
10,
conditions=AssayConditions(pH=7.0),
system=ProteinLigandComplex(components=[ligand, protein]),
)
measurement
[22]:
<PercentageDisplacementMeasurement values=[10] conditions=<AssayConditions pH=7.0> system=<ProteinLigandComplex with 2 components (<Ligand name=propane>, <Protein name=NTRK1>)>>
DatasetProviders¶
DatasetProviders are essentially a list of Measurements, which can be used for machine learning experiments. Featurizers can be passed to allow a featurization of all available Systems. Currently, KinoML is shipped with DatasetProviders for PKIS2 and ChEMBL datasets allowing quick experiment design.
[23]:
from kinoml.datasets.chembl import ChEMBLDatasetProvider
from kinoml.datasets.pkis2 import PKIS2DatasetProvider
[24]:
# load data points given by the PKIS2 publication (https://doi.org/10.1371/journal.pone.0181585)
pkis2 = PKIS2DatasetProvider.from_source()
print(pkis2)
<PKIS2DatasetProvider with 261870 PercentageDisplacementMeasurement measurements and 261870 systems (Ligand=640, KLIFSKinase=406)>
[25]:
# load curated ChEMBL data points available at https://github.com/openkinome/kinodata
# here the more general "Protein" object will be used instead of the default "KLIFSKinase"
# also protein objects will be initialized with the MDAnalysis toolkit
chembl = ChEMBLDatasetProvider.from_source(
path_or_url="https://github.com/openkinome/datascripts/releases/download/v0.3/activities-chembl29_v0.3.zip",
measurement_types=("pIC50", "pKi", "pKd"),
protein_type="Protein",
toolkit="MDAnalysis",
)
chembl
[25]:
<ChEMBLDatasetProvider with 190469 measurements (pIC50Measurement=160703, pKiMeasurement=15653, pKdMeasurement=14113), and 188032 systems (Protein=462, Ligand=115207)>
[26]:
# loading a smaller sample allows rapid testing
# loading now with default "KLIFSKinase" protein object
chembl = ChEMBLDatasetProvider.from_source(
path_or_url="https://github.com/openkinome/datascripts/releases/download/v0.3/activities-chembl29_v0.3.zip",
measurement_types=["pKi"],
sample=100,
)
chembl
[26]:
<ChEMBLDatasetProvider with 100 measurements (pKiMeasurement=100), and 100 systems (KLIFSKinase=38, Ligand=100)>
[27]:
%%capture --no-display
# upper statement to hide warnings
# all systems will be successfully featurized
chembl.featurize(MorganFingerprintFeaturizer())
chembl
[27]:
<ChEMBLDatasetProvider with 100 measurements (pKiMeasurement=100), and 100 systems (KLIFSKinase=38, Ligand=100)>
[28]:
from kinoml.features.protein import OneHotEncodedSequenceFeaturizer
[29]:
# not all systems maybe featurizable and will be removed, e.g. erroneous SMILES
# here certain ChEMBL data points are for kinases that are not available in KLIFS
chembl.featurize(OneHotEncodedSequenceFeaturizer(sequence_type="klifs_kinase"))
chembl
There were 3 systems that could not be featurized!
[29]:
<ChEMBLDatasetProvider with 97 measurements (pKiMeasurement=97), and 97 systems (KLIFSKinase=37, Ligand=97)>